Diffuse Intrinsic Pontine Glioma - (DIPG) is a highly malignant brain tumour (cancer) located in the 'PONS' or pontine area of the brain stem, almost exclusively affecting children.
It is a type of 'DMG' and may also be referred to as a diffuse midline glioma.
There is currently NO cure.
The brain stem is the bottom most portion of the brain, connecting the cerebrum with the spinal cord. The majority of brain stem tumours occur in the 'pons' this sits atop the delicate brain stem, an area that controls life-essential functions: breathing, swallowing, balance, facial expression. The growing tumour mass devastates all of these functions, they are diffusely infiltrating (they grow amidst the nerves), and therefore are not able to be surgically removed due to location and damage to healthy tissue.
Diffuse Midline Glioma (DMG)
Following the 2021 revision to WHO classification, paediatric diffuse high-grade gliomas now fall into 4 groups:
- Diffuse Midline Glioma, H3 K27 - Altered
- Diffuse Hemispheric Glioma, H3 G34 - Mutant
- Diffuse High Grade Glioma, H3 wildtype, IDH wildtype
- Infant type Hemispheric Glioma
Pontine (pons) Glioma
Pontine gliomas are highly malignant (cancerous) and aggressive tumours that originate in the pons. Diffuse Intrinsic Pontine Glioma (DIPG) is a hugely difficult to treat brain tumour . They are glial tumours, meaning they arise from the brain's glial tissue—tissue made up of cells that help support and protect the brain's neurons.They are 'primary' meaning they have not spread but can grow very quickly...DIPG children typically experience one month or less of symptoms before they are diagnosed and then worsen rapidly, related to the growing tumour causing compression or dysfunction of anatomic structures in and near the pons.
DIPG affects children almost exclusively who are typically between the ages of 4 and 11, there is no evidence to say that girls are affected more than boys or vice versa. It is not hereditary or able to be 'passed on'
DIPG commonly causes patients to experience double vision, reduced eye movement, facial weakness or asymmetry, and arm and leg weakness. They also have problems with walking, coordination, speech, chewing, and swallowing.
As the tumour progresses, it also interferes with breathing and heartbeat, which ultimately results in the child’s death. DIPG is thought to be responsible for 80% of all of the brain tumour death that occurs in children.
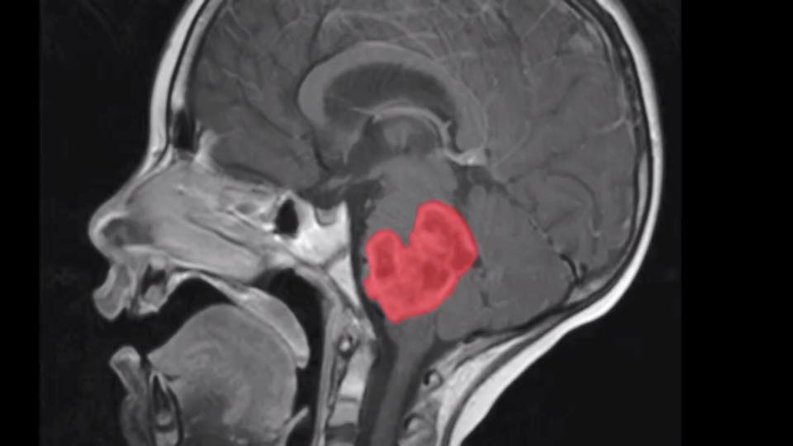
IF a biopsy can be carried out they will almost always be found to be ' high grade(4)' gliomas and may also be classified under new WHO coding as Diffuse Midline Glioma, H3 K27 - Altered .
Any tumour harbouring the H3 K27 mutation in a midline location, may in fact be termed a 'DMG' when 'DIPG' references the anatomical position specific to a 'pontine' location as the tumour origination site in the brain stem.
So not all 'diffuse midline gliomas' are DIPG's ( eg those originating in the thalamus) but in either instance sadly the prognosis is poor. (Scan above is Abbie's post radiation)
Who gets pontine glioma?
On average 40 children a year develop pontine glioma in the UK. This is around 10-15% of all childhood brain tumours that occur in UK children. They rarely occur in adults. Like most brain tumours, the cause of pontine gliomas is unknown
What is the prognosis for children diagnosed?
Medical advances in the past 40 years have greatly improved the survival rates for children diagnosed with most types of cancer. For some cancers, the medical advances have been extraordinary. For example, the survival rate for children with acute lymphocytic leukemia (ALL) has increased from less than 10 percent in the 1960s to nearly 90 percent today. Overall, the survival rate for children with cancer is around 83% percent.
But these medical advances have done nothing for children with DIPG. Brain tumours remain the most common cause of cancer-related death in children, and DIPG is the leading cause of death from paediatric brain tumours. A child diagnosed with DIPG today faces the same prognosis as a child diagnosed 40 years ago. There is still no effective treatment and no chance of survival. Only 10% of children with DIPG survive for 2 years following their diagnosis, and less than 1% survive for 5 years. The median survival time is 9 months from diagnosis.
Cytogenetic Characteristics of Diffuse Intrinsic Pontine Gliomas
The genomic characteristics of DIPGs appear to differ from those of most other pediatric high-grade gliomas and from those of adult high-grade gliomas. A number of chromosomal and genomic abnormalities have been reported for DIPG, including the following:
Histone H3 genes: Groundbreaking work uncovered novel point mutations of highly conserved chromatin remodeling genes H3F3A, HIST1H3B and HIST1H3C, all resulting in a lysine to methionine substitution at residue 27 (K27M), which have since been reported in 70-96% of DIPG specimens. These same mutations are observed in pediatric high-grade gliomas at other midline locations but are uncommon in cortical pediatric high-grade gliomas and in adult high-grade gliomas.
The recent WHO re-classification (2021) includes DIPG in the group term of Diffuse Midline Glioma, H3 K27 - Altered referencing any mid-line tumour with the H3 K27M mutation as a phenotypically and molecularly defined set of tumors ...DIPG remains anotomically dependent on location and exact pontine region , elsewhere there may well be other mutations and deletions to consider regarding any targeted treatment.
Activin A receptor, type I (ACVR1) gene: Approximately 20% of DIPG cases have activating mutations in the ACVR1 gene. Patients harbouring ACVR1 mutations are predominantly female (approx. 2:1), and have a younger age of onset (approx. 5 years) and longer overall survival time (approx. 15 months) compared with wild-type tumours. ACVR1 mutations also strongly co-segregate with K27M mutations in the gene encoding histone H3.1 (HIST1H3B), which themselves are now reported to represent an accumulated 22% of DIPG with most occurring concurrently with H3.3 mutations. Germline mutations in ACVR1 also cause the autosomal dominant syndrome fibrodysplasia ossificans progressiva (FOP), although there is no cancer predisposition in FOP.
Receptor tyrosine kinase amplification: PDGFRA amplification occurs in approximately 30% of cases, with lower rates of amplification observed for some other receptor tyrosine kinases (e.g., MET and IGF1R).
TP53 deletion: DIPG tumours commonly show deletion of the TP53 gene on chromosome 17p. Additionally, TP53 is commonly mutated in DIPG tumours, particularly those with histone H3 gene mutations. Aneuploidy is commonly observed in cases with TP53 mutations.
The gene expression profile of DIPG differs from that of non–brain stem pediatric high-grade gliomas, further supporting a distinctive biology for this subset of pediatric gliomas.